
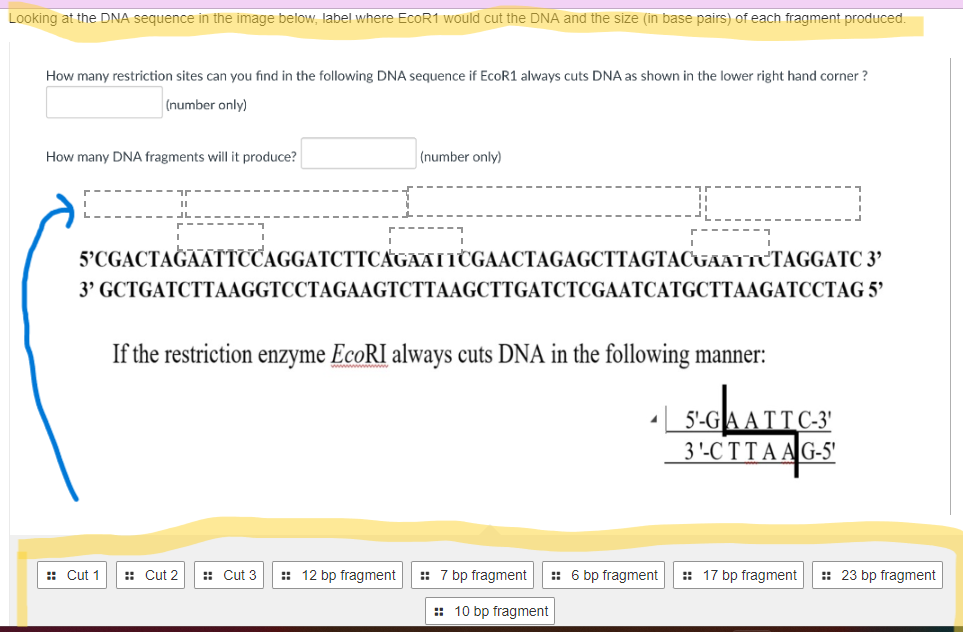
Restriction endonucleases, ubiquitous tools in molecular biology, cleave DNA at specific recognition sequences, often palindromic. The precise localization of these restriction sites within a DNA molecule is crucial for various applications, including DNA cloning, genetic mapping, and Southern blotting. Identifying these sites, therefore, is a foundational skill for any molecular biologist. This comprehensive guide elucidates the multifaceted approaches to locating restriction sites within a given DNA sequence.
I. Understanding Restriction Enzyme Specificity
The cornerstone of restriction site identification lies in comprehending the sequence specificity of each restriction enzyme. These enzymes, sourced from bacteria as a defense mechanism against bacteriophages, recognize short DNA sequences, typically 4-8 base pairs in length. Some recognize degenerate sequences, allowing for cleavage at multiple related, but not identical, sites. This degeneracy arises from the presence of ambiguous bases in the recognition sequence, represented by IUPAC nucleotide codes. Each enzyme possesses a unique recognition sequence, and consequently, a unique cleavage pattern.
A. Navigating Restriction Enzyme Databases
Several publicly accessible databases serve as invaluable resources for information pertaining to restriction enzymes. These databases, such as REBASE (Restriction Enzyme Database) and those hosted by enzyme vendors like New England Biolabs (NEB), provide comprehensive details about each enzyme. Information includes the recognition sequence, cleavage site (which might be within the recognition sequence or outside of it), optimal reaction conditions, commercial availability, and any isoschizomers. Isoschizomers are enzymes that recognize the same sequence but may exhibit different cleavage patterns or be sensitive to different methylation states.
B. Deciphering IUPAC Nucleotide Codes
Restriction enzyme recognition sequences are often represented using IUPAC nucleotide codes. Familiarity with these codes is essential for accurate site identification. For example, ‘R’ represents a purine (A or G), ‘Y’ represents a pyrimidine (C or T), ‘N’ represents any base (A, G, C, or T), and ‘W’ represents A or T. Understanding these ambiguities is paramount. Failure to account for them can lead to overlooking potential restriction sites.
II. In Silico Restriction Site Mapping
The advent of bioinformatics has revolutionized restriction site identification. In silico mapping, utilizing computational tools, provides a rapid and accurate method for predicting restriction sites within a DNA sequence. Several software packages and online tools are available, each offering a unique set of features and functionalities.
A. Utilizing Web-Based Restriction Mapping Tools
Numerous web-based tools offer user-friendly interfaces for restriction site mapping. These tools typically require the user to input a DNA sequence (in FASTA format or plain text) and select the desired restriction enzymes. The software then analyzes the sequence and generates a map indicating the positions of all restriction sites for the chosen enzymes. Examples include NEBcutter V3.0 (provided by New England Biolabs) and RestrictionMapper. These tools often offer advanced features, such as the ability to filter results based on the number of cuts, the size of the resulting fragments, and the presence of methylation-sensitive enzymes. They can also display the results graphically, enhancing visualization.
B. Employing Dedicated Bioinformatics Software
For more sophisticated analyses, dedicated bioinformatics software packages such as Geneious Prime, CLC Main Workbench, and SnapGene offer comprehensive restriction mapping capabilities. These programs often integrate restriction mapping with other molecular biology tools, such as sequence alignment, primer design, and phylogenetic analysis. They generally support larger datasets and provide greater customization options, allowing for fine-tuning of the analysis parameters. Many also incorporate features to simulate gel electrophoresis, predicting the size and distribution of DNA fragments generated by restriction enzyme digestion.
C. Scripting for Automation and Customization
For large-scale analyses or customized workflows, scripting languages like Python or R can be employed. Biopython, a Python library for bioinformatics, provides modules for sequence manipulation and restriction enzyme analysis. Similarly, R packages like ‘Biostrings’ offer functionalities for pattern matching and sequence analysis. These scripting approaches allow for automation of the restriction mapping process and integration with other bioinformatics pipelines, enabling efficient handling of large genomic datasets.
III. Visual Inspection and Manual Analysis
While in silico methods are efficient, visual inspection of the DNA sequence can be beneficial, especially for educational purposes or when dealing with short sequences. This approach involves manually searching for the recognition sequences of the desired restriction enzymes within the DNA sequence.
A. Identifying Palindromic Sequences
Many restriction enzymes recognize palindromic sequences, where the sequence reads the same forward and backward on complementary strands. Identifying potential restriction sites requires recognizing these palindromic patterns within the DNA sequence. This manual search can be tedious but reinforces understanding of restriction enzyme specificity.
B. Accounting for Degeneracy and Ambiguity
When manually searching for restriction sites, it is crucial to account for any degenerate bases or ambiguities in the recognition sequence. The IUPAC nucleotide codes must be carefully considered to ensure that all potential sites are identified. Overlooking these ambiguous positions can lead to inaccurate mapping.
IV. Experimental Validation of Restriction Sites
While in silico and manual methods provide predictions of restriction sites, experimental validation is often necessary to confirm the presence and accessibility of these sites. This involves digesting the DNA molecule with the restriction enzyme and analyzing the resulting fragments using gel electrophoresis.
A. Restriction Digestion and Gel Electrophoresis
Restriction digestion involves incubating the DNA molecule with the appropriate restriction enzyme under optimal reaction conditions. The enzyme cleaves the DNA at the predicted restriction sites, generating a set of DNA fragments. These fragments are then separated by size using agarose gel electrophoresis. The resulting banding pattern can be compared to the predicted fragment sizes to confirm the presence and location of the restriction sites.
B. Optimizing Reaction Conditions
Optimal reaction conditions are crucial for efficient restriction digestion. Factors such as temperature, pH, salt concentration, and the presence of cofactors can significantly affect enzyme activity. It’s important to consult the enzyme manufacturer’s recommendations to ensure optimal digestion.
C. Addressing Potential Challenges
Several factors can complicate experimental validation. Incomplete digestion, due to enzyme inactivation or inhibited access to the DNA, can result in unexpected banding patterns. DNA methylation can also interfere with restriction enzyme activity, as some enzymes are sensitive to methylation at their recognition sites. Furthermore, DNA secondary structures can hinder enzyme access. Addressing these challenges may require optimizing digestion conditions, using methylation-insensitive enzymes, or denaturing the DNA prior to digestion.
In conclusion, identifying restriction sites in a DNA sequence involves a combination of understanding enzyme specificity, utilizing in silico tools, and, in some cases, performing experimental validation. Mastering these techniques is crucial for anyone working in molecular biology, enabling efficient manipulation and analysis of DNA molecules. The ability to accurately locate and utilize restriction sites forms the foundation for a wide range of molecular biology applications.







Leave a Comment